


RMSD Analysis in CPPTRAJ
Formerly known as "Tutorial A1: Setting up an Advanced System
By Daniel R. Roe, July 2014
TABLE OF CONTENTS
IntroductionCalculating RMSD
Loading a Reference Structure
Calculating RMSD to a Reference
RMSD to Reference with Different Topology
Associated Files
Introduction
This tutorial will cover one of the most basic types of analysis performed after MD simulations: coordinate root-mean-squared deviation (RMSD). It will also cover 'tagging' loaded topology and reference files in CPPTRAJ.
RMSD measures the deviation of a target set of coordinates (i.e. a structure) to a reference set of coordinates, with RMSD=0.0 indicating a perfect overlap. RMSD is defined as:
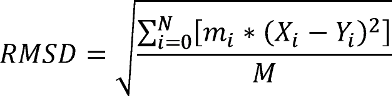
When calculating RMSD of a target to reference structure, there are two
very important requirements:
Pre-requisites
This tutorial assumes that you have installed and tested AmberTools, and that you have completed the first CPPTRAJ tutorial. In addition, xmgrace will be required to view some of the output data. A molecular graphics viewing program (such as VMD or Chimera) is recommended for viewing structures/trajectories.
Throughout this tutorial a short example trajectory of the beta-hairpin trpzip2 will be used. The trajectory is in NetCDF format, which is faster to process, more compact, higher precision, and more robust than the ASCII format. NetCDF is enabled by default in Amber, but if you find that your CPPTRAJ cannot read this trajectory please contact the Amber mailing list for help. The trajectory, associated topology, and other files can be downloaded here:
- trpzip2.gb.nc ¦ MD5 sum: 059435cfe1fcd57c327d46711da9ceff
- trpzip2.ff10.mbondi.parm7 ¦ MD5 sum: 140044a45c683612da3431dfd0697e22
- trpzip2.1LE1.1.rst7 ¦ MD5 sum: 9d4a64e65c77e9833ff38a424b4669d0
- trpzip2.1LE1.10.rst7 ¦ MD5 sum: 6ea49fa08bae5fd92fb4246ee7ec34ca
- 2GB1.pdb ¦ MD5 sum: 69fd269e026f25aef2a5e94654296ae5
For more detailed information on CPPTRAJ, see here:
Calculating RMSD
To start CPPTRAJ, type 'cpptraj' from the command line:
[user@computer ~]$ cpptraj
CPPTRAJ: Trajectory Analysis. V14.05
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
>
Load the topology and trajectory file:
> parm trpzip2.ff10.mbondi.parm7 Reading 'trpzip2.ff10.mbondi.parm7' as Amber Topology > trajin trpzip2.gb.nc Reading 'trpzip2.gb.nc' as Amber NetCDF
Specify the RMSD command:
> rms ToFirst :1-13&!@H= first out rmsd1.agr mass
RMSD: (:1-13&!@H*), reference is first frame (:1-13&!@H*), with fitting, mass-weighted.
In this case we are calculating mass-weighted RMSD, saving to a data set named 'ToFirst', using all non-hydrogen atoms in residues 1 to 13, using the first frame as reference, and writing to a file named 'rmsd1.agr' (which will be written in xmgrace format due to the .agr extension). Note that by default the 'rms' command in CPPTRAJ calculates the best-fit RMSD, which means each structure is rotated and translated so as to minimize the RMSD to the reference structure (in this case the first frame). As such, the 'rms' command modifies coordinates for all subsequent commands unless 'nofit' is specified (more on that later).
Start the trajectory processing by typing 'run'. Once the run has completed, if xmgrace is installed you can view the output right from the CPPTRAJ command line:
> xmgrace rmsd1.agr
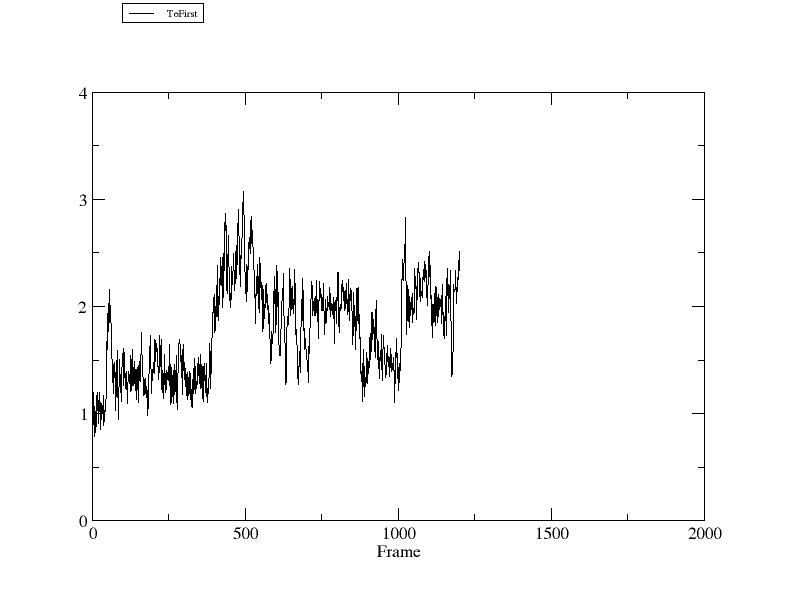
In this figure the X axis is the frame number and the Y axis is coordinate RMSD to the first frame (in Angstroms).
Loading a Reference Structure
While the RMSD to the first frame can be a useful indicator of how much the structure deviates from its initial configuration, typically one is also interested in calculating deviation from a specific reference structure, such as one obtained via an experimental method (e.g. X-ray crystallography or NMR). The PDB entry for trpzip2 studied via NMR is 1LE1. The Amber restart file 'trpzip2.1LE1.1.rst7' contains the coordinates for the first member of the NMR ensemble. This structure can be loaded as a reference by using the 'reference' command.
> reference trpzip2.1LE1.1.rst7 Reading 'trpzip2.1LE1.1.rst7' as Amber Restart 'trpzip2.1LE1.1.rst7' is an AMBER restart file, no velocities, Parm trpzip2.ff10.mbondi.parm7 (reading 1 of 1)
Similarly, the Amber restart file 'trpzip2.1LE1.10.rst7' contains coordinates for the 10th member of the NMR ensemble:
> reference trpzip2.1LE1.10.rst7 [tenth_member] Reading 'trpzip2.1LE1.10.rst7' as Amber Restart 'trpzip2.1LE1.10.rst7' is an AMBER restart file, no velocities, Parm trpzip2.ff10.mbondi.parm7 (reading 1 of 1)
In this case we have tagged the reference with '[tenth_member]'. In cpptraj, both reference structures and topology files can be tagged when they are loaded by providing a bracket-enclosed name, i.e. [<name>]. The files can then be referred to by file name, by index (i.e. the order they were loaded starting from 0), or by the tag.
Any trajectory file that CPPTRAJ can normally read can be used as a reference structure by specifying the desired frame number, or 'lastframe' to explicitly select the final trajectory frame. For example, say we wanted to use the final frame of the trajectory as a reference structure. We can use the 'lastframe' keyword and tag it [last] like so:
> reference trpzip2.gb.nc lastframe [last] Reading 'trpzip2.gb.nc' as Amber NetCDF 'trpzip2.gb.nc' is a NetCDF AMBER trajectory, Parm trpzip2.ff10.mbondi.parm7 (reading 1 of 1201)
The 'list' command can provide information on the currently loaded reference structures:
> list ref
REFERENCE FRAMES (3 total):
0: 'trpzip2.1LE1.1.rst7', frame 1
1: [tenth_member] 'trpzip2.1LE1.10.rst7', frame 1
2: [last] 'trpzip2.gb.nc', frame 1201
Active reference frame for masks is 0
Here we see the three reference structures that have been loaded, along with their indices, names, and tags (if a tag was specified).
Calculating RMSD to a Reference
Now that we have loaded some reference structures, we can calculate the RMSD to each. There are three keywords that can be used to select reference structures:
- reference: Use the first loaded reference structure.
- refindex <#>: Use reference index number <#> (so 'reference' is like 'refindex 0').
- ref <name | tag>: Use reference specified by file name or tag.
> rms ToMember1 :1-13&!@H= reference out rmsd2.agr
Mask [:1-13&!@H*] corresponds to 116 atoms.
RMSD: (:1-13&!@H*), reference is reference frame trpzip2.1LE1.1.rst7 (:1-13&!@H*), with fitting.
> rms ToMember10 :1-13&!@H= refindex 1 out rmsd2.agr
Mask [:1-13&!@H*] corresponds to 116 atoms.
RMSD: (:1-13&!@H*), reference is reference frame trpzip2.1LE1.10.rst7 (:1-13&!@H*), with fitting.
> rms ToLast :1-13&!@H= ref [last] out rmsd2.agr
Mask [:1-13&!@H*] corresponds to 116 atoms.
RMSD: (:1-13&!@H*), reference is reference frame trpzip2.gb.nc (:1-13&!@H*), with fitting.
Start the trajectory processing by typing 'run'. Once the run has completed, if xmgrace is installed you can view the output right from the CPPTRAJ command line:
> xmgrace rmsd2.agr
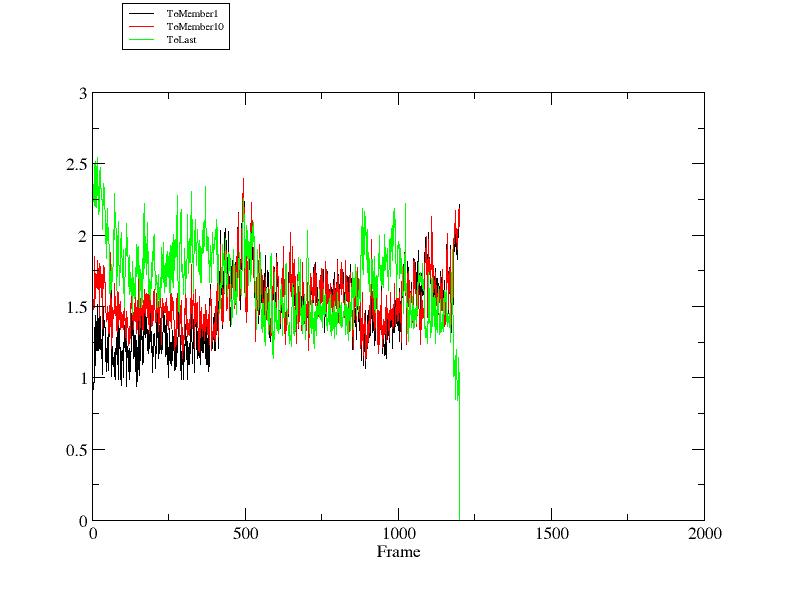
Here we can see that the trajectory starts out slightly closer to member 1 than member 10.
RMSD to Reference with Different Topology
It is often useful to look at the RMSD between structures which may not have the same topology. For example, a related hairpin structure is that of the B1 IgG-binding domain of protein G (GB1, PDB ID: 2GB1), which has a similar arrangement of hydrophobic residues in its strands. However, it has a different turn motif as well as two additional residues in the turn region. One might be interested with how closely the strands of trpzip2 resemble those of GB1, or want to align the structure of trpzip2 with the GB1 hairpin. This can be done by loading GB1 as a reference structure and then choosing a target mask for trpzip2 and a reference mask for GB1 so that they overlap. Examination of the two structures reveals that residues 1 to 6 and 7 to 12 of trpzip2 overlap with residues 42 to 47 and 50 to 55 of GB1.
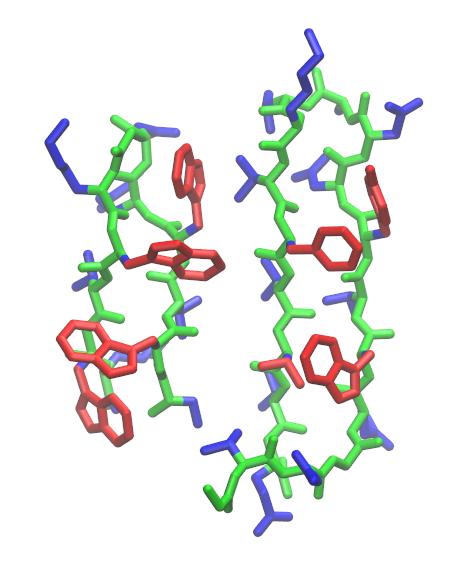
Since GB1 has a different topology than trpzip2, we need to load it as a new topology.
> parm 2GB1.pdb Reading '2GB1.pdb' as PDB File 2GB1.pdb: determining bond info from distances. Warning: 2GB1.pdb: Determining default bond distances from element types.
Note that since PDB files do not contain connectivity, CPPTRAJ automatically attempts to determine connectivity based on inter-atomic distances and element types.
We can now load 2GB1 as a reference structure. However, since 2GB1 is not using the first topology loaded, we need to specify the topology to use. Similar to reference structures, topologies can be specified by index, name, or tag.
- parmindex <#>: Use topology index number <#>.
- parm <name | tag>: Use topology specified by file name or tag.
> reference 2GB1.pdb parm 2GB1.pdb [GB1] Reading '2GB1.pdb' as PDB '2GB1.pdb' is a PDB file, Parm 2GB1.pdb (reading 1 of 1)
We now specify the 'rms' command, but with slight differences to the previous commands. First, since the residues are mostly different between the two strands, and the atom ordering in the PDB may be different than what is in our topology, we will only use alpha carbons. Second, we need to specify two masks: one which describes the atoms to select in trpzip2, and one that describes the atoms to select in GB1.
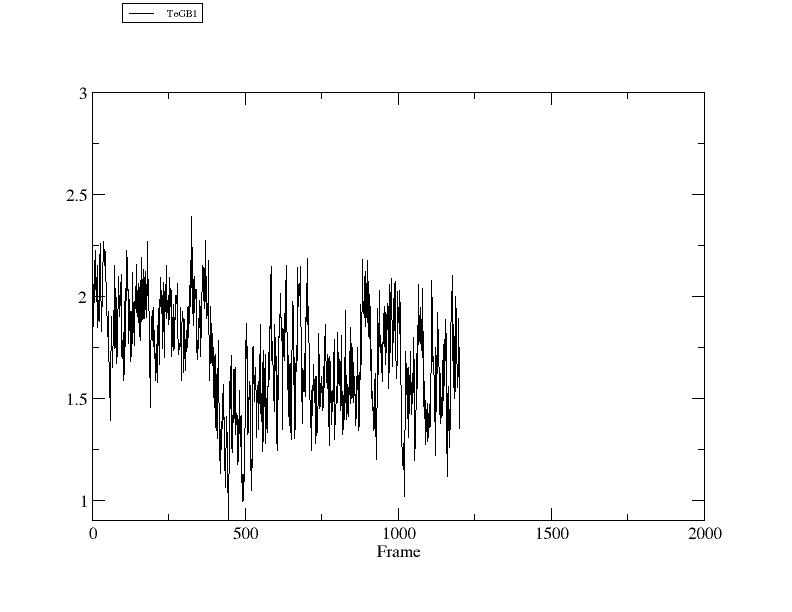
> rms ToGB1 ref [GB1] :1-12@CA :42-47,50-55@CA out rmsd3.agr
Mask [:42-47,50-55@CA] corresponds to 12 atoms.
RMSD: (:1-12@CA), reference is reference frame 2GB1.pdb (:42-47,50-55@CA), with fitting.
We will also tell CPPTRAJ to write out the first frame of trpzip2 after it has been RMS-best-fit, using the 'onlyframes' keyword.
> trajout trpzip2.overlap.mol2 onlyframes 1 Writing 'trpzip2.overlap.mol2' as Mol2 Saving frames 1
Type 'run' to process the trajectory. You can see in the output that the number of target atoms selected during trajectory processing matches the number of atoms selected for the reference.
ACTION SETUP FOR PARM 'trpzip2.ff10.mbondi.parm7' (1 actions): 0: [rms ToGB1 ref [GB1] :1-12@CA :42-47,50-55@CA out rmsd3.agr] Mask [:1-12@CA] corresponds to 12 atoms.
Once the run has completed, if xmgrace is installed you can view the output right from the CPPTRAJ command line:
> xmgrace rmsd3.agr
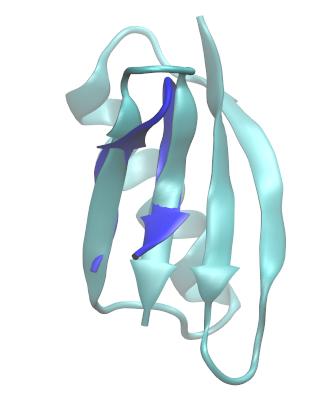
If you visualize the output structure, you will see that the trpzip2 coordinates have been best-fit to the specified strands in GB1.
Associated Files
These files can be used to check your output.
rmsd1.inrmsd1.agr
rmsd2.in
rmsd2.agr
rmsd3.in
rmsd3.agr
trpzip2.overlap.mol2
Copyright Daniel R. Roe, 2014