


Copyright (C) Pengfei Li & Kenneth M. Merz Jr. 2015
Modeling a Magnesium-DNA System using the 12-6-4 LJ-Type Nonbonded Model
Friendly remind that only pmemd.cuda in Amber18 or higher version support the 12-6-4 simulations. While pmemd.cuda in the former versions does not support the 12-6-4 simulations. Please refer the AMBER manual about the "pmemd and pmemd.amoeba"(or just "pmemd")->"GPU Accelerated PMEMD"-> "Supported Features" section.
Here we treat a DNA fragment (PDB ID: 1D23) as an example for building the 12-6-4 LJ-type nonbonded model in the AMBER force field. There are two options: A) Using the default parameters; B) Using the fine-tuned parameters from Panteva et al. (the m12-6-4 parameter set).
Example A: Using Default Parameters
The first example is using default parameters. The parameters and combining rule are from Li and Merz.[1] In the work of Panteva et al. they found the 12-6-4 model with SPC/E water performs the best among the nonbonded models investigated. [2] Here we also use the SPC/E water model in our example.
A1. Clean the PDB file (effort: several minutes):
Here is the original PDB file: 1D23.pdb. There are no hydrogen atoms in the file. tleap will automatically add them in next step. Users can also use webserver H++ to add hydrogen atoms, which is a more accurate way to determine the protonation state of residues, please check the MCPB.py tutorial for more details.
Here we perform the command:
awk '$1=="ATOM" || $1=="HETATM" || $1=="TER" || $1=="END"' 1D23.pdb > 1D23_clean.pdb
Here is the PDB file after cleaning: 1D23_clean.pdb.
A2. Generate the topology and coordinate files (effort: several minutes):
In this step we need to create an input file manually. Here is the input file: tleap_1264.in.
Below is the explanation of the input file: (Reminder: please load the 12-6-4 parameter set for the atomic ions.)
source leaprc.DNA.OL15 #Source the OL15 force field for DNA
source leaprc.water.spce #Source the water model and related metal ion force fields
loadamberparams frcmod.ions234lm_1264_spce #Load the 12-6-4 parameter set for +2, +3, and +4 metal ions for SPC/E water model
mol = loadpdb 1D23_clean.pdb #Load the PDB file
solvatebox mol SPCBOX 10.0 #Add water box
loadamberparams frcmod.spce #Load frcmod file for SPC/E water model
addions mol MG 0 #Neutralize the system with Mg2+ ions, here MG is the unit name
savepdb mol 1D23_solv.pdb #Save PDB file
saveamberparm mol 1D23_solv.prmtop 1D23_solv.inpcrd #Save topology and coordiante files
quit #Quit the program
The atomic_ions.lib file, which contains the library of atomic ions, is loaded when we source the leaprc.water.spce file. There are Mg2+ unit inside the atomic_ions.lib file therefore we can perform "addions mol MG 0" command in the input file to neutralize the system with Mg2+ ions. If the total charge of the system is positive, we can use "addions mol Cl- 0" command in the input file to neutralize the system with Cl- ions while the Cl- unit is also contained in the atomic_ions.lib file. If you create a lib file for metal ion(s) by yourself, please use a different name from the "atomic_ions.lib" file, preventing conflict with the existing file. The above example is for AMBER 2016. Note that the names of leaprc files for different force fields and frcmod files for ions may change between different AMBER versions, hence some commands in above input file may need to be changed or deleted. Users can check the Specifying which force field you want in LEaP and Ions sections in the Molecular mechanics force fields chapter in the corresponding AMBER manual and $AMBERHOME/dat/leap/cmd/ directory for more details. We can run the following command to generate the topology and coordinate files:
tleap -s -f tleap_1264.in > tleap_1264.out
A3. Add C4 terms into the topology file (effort: several minutes):
In this step we need to use ParmEd to add C4 terms to the end of the topology file. Users can check the add12_6_4 command for the ParmEd software in the Amber manual for more details. It supports adding C4 terms for different kind of ions (if you have more than one kind of ions using the 12-6-4 model), for which you can specify in the amber mask in the add12_6_4 command. Here is the input file for ParmEd: 1264_parmed.in
We can run the command:
parmed -i 1264_parmed.in -p 1D23_solv.prmtop
We can see the "%FLAG LENNARD_JONES_CCOEF" terms in the end of the 1D23_solv_1264.prmtop file, which are the C4 terms for the system.
A4. Check parameters in the topology file:
Users can use the "printDetails" command in ParmEd to check the detailed information about the Mg2+ ions. Moreover, users can use the "printLJMatrix" command in ParmEd to check the C12, C6 and C4 parameters related to the Mg2+ ions (C4 parameters are only checkable by using ParmEd in Amber16 or higher version). An example has shown in example shown in the 12-6 nonbonded model tutorial.
Afterwards you can use the new 1D23_solv_1264.prmtop and 1D23_solv_1264.inpcrd files to perform the simulations now. Make sure you added "lj1264=1" in the control list of the input files if you are using Amber14 or lower version (please check lj1264 in manual for more details).
Example B: Using Fine-tuned Parameters from Panteva et al. (the m12-6-4 Parameter Set)
(This part of tutorial needs AmberTools16 or Higher)
Panteva et al. have employed the 12-6-4 model from Li and Merz and fine-tuned the C4 terms between several divalent metal ions (Mg2+, Mn2+, Zn2+, and Cd2+) and nucleic acid systems in TIP4PEW water box.[3] The C4 term between metal ion and water was kept from the original value so the excellent representation of ion-water interaction has not been changed. The new parameter set (referred as the m12-6-4 parameter set) has better balance of interactions between metal ion and nucleic acids. Here we use the same DNA system (PDB ID: 1D23) as an example and take advantage of the files we obtained in the Example A above.
B1. Prepare the original prmtop and inpcrd files:
In order to use the new parameters we need to use a new tleap input file: tleap_1264_tip4pew.in
tleap -s -f tleap_1264_tip4pew.in > tleap_1264_tip4pew.out
The names of leaprc files for different force fields and frcmod files for ions may change between different AMBER versions, hence some commands in above input file may need to be changed or deleted. Users can check the Specifying which force field you want in LEaP and Ions sections in the Molecular mechanics force fields chapter in the corresponding AMBER manual and $AMBERHOME/dat/leap/cmd/ directory for more details.
B2. Add C4 terms into the topology file (use Panteva fine-turned parameters, effort: several minutes):
Here we need to use a new ParmEd input file: parmed_1264_na.in
Here is the explaination for the ParmEd input file content:
setOverwrite True #Set overwrite output file
change AMBER_ATOM_TYPE :A*,DA*@N7 NAMG #Change atom type of Adenine N7 atom to NAMG
change AMBER_ATOM_TYPE :G*,DG*@N7 NGMG #Change atom type of Adenine N7 atom to NGMG
change AMBER_ATOM_TYPE :*@OP* OPMG #Change atom type of backbone phosphate oxygen to OPMG
addLJType @%NAMG #Add a new LJ type for Adenine N7 atom
addLJType @%NGMG #Add a new LJ type for Guanine N7 atom
addLJType @%OPMG #Add a new LJ type for backbone phosphate oxygen
add12_6_4 :MG watermodel TIP4PEW #Add C4 terms
printLJMatrix :MG #Print out the LJ matrix to check whether the adding C4 terms are right
outparm 1D23_1264_na_tip4pew.prmtop 1D23_1264_na_tip4pew.inpcrd #Output new prmtop and inpcrd files
We perform the command:
parmed -i parmed_1264_na.in -p 1D23_tip4pew.prmtop -c 1D23_tip4pew.inpcrd
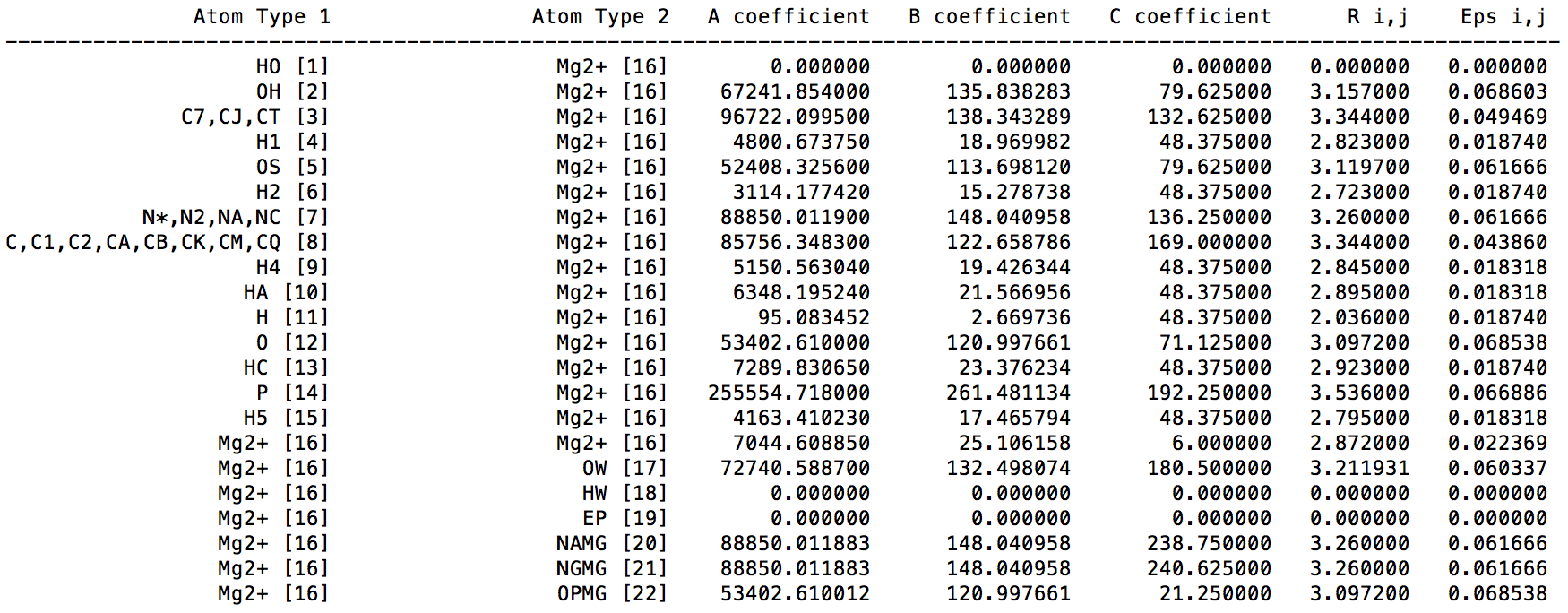
In the ParmEd output, we can see following content for "printLJMatrix" command:
The A, B, and C coefficients are C12, C6, and C4 terms respectively. We can see the C4 term between Mg2+ and OW is 180.5, which is constent with the original paper (in Table 4 of ref 1). [1]
Afterwards you can use the new 1D23_solv_1264_na_tip4pew.prmtop and 1D23_solv_1264_na_tip4pew.inpcrd files to perform the simulations now. Make sure you added "lj1264=1" in the control list of the input files if you are using Amber14 or lower version (please check lj1264 in manual for more details).
References:
[1] Pengfei Li and Kenneth M. Merz, Jr. "Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions", J. Chem. Theory Comput., 2014, 10, 289-297
[2] Maria T. Panteva, George M. Giambasu, Darrin M. York "Comparison of Structural, Thermodynamic, Kinetic and Mass Transport Properties of Mg2+ Ion Models Commonly Used in Biomolecular Simulations", J. Comput. Chem., 2015, 13, 970-982
[3] Maria T. Panteva, George M. Giambasu, Darrin M. York "Force Field for Mg2+, Mn2+, Zn2+ and Cd2+ Ions That Have Balanced Interactions with Nucleic Acids", J. Phys. Chem. B, 2015, 119, 15460-15470