Using the Amber force field in NAMD
George M. Giambaşu and David A. Case
1 Why would you want to do this?
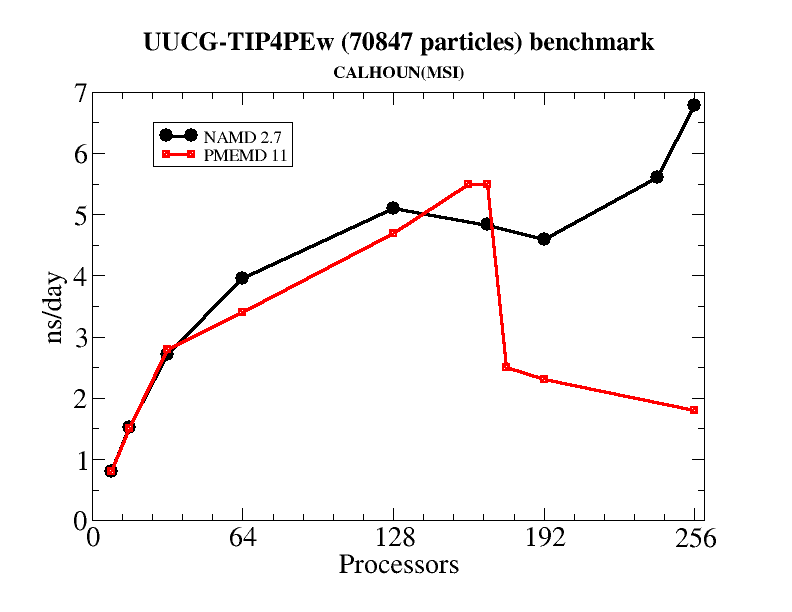
Generally, the pmemd module of Amber is the recommended path for carrying out parallel MD simulations. At fairly low processor counts, say up to 8-160 threads (for a modern, infiniband cluster) NAMD 2.7 and pmemd are rather similar in performance (see the table below for an example.) But at higher numbers of threads, it is typical for the parallel scaling in pmemd to degrade signficantly, such that adding more threads may even slow things down. NAMD, on the other hand, continues to scale well at 256 threads and beyond. Hence, if you have access to large numbers of nodes in a well-connected cluster, and want to use them for a single simulation, NAMD can be a good option.
2 Getting compatible settings
| NAMD2.7 settings | Note | Corresponding Amber11 settings |
| Force Field Settings | ||
| amber on | Use Amber FF, expect to read a PARM7 file. | default in amber |
| switching off | Do not use switching | default in amber |
| exclude scaled1-4 | 1-4 interactions are scaled by scnb and 1-4scaling, 1-3 interactions are ignored. | default in amber |
| 1-4scaling 0.833333333 | Scaling factor for 1-4 electrostatic interaction(0.833333333=1.0/1.2). | scee=1.2 |
| scnb 2.0 | 1-4 vdW interactions are divided by scnb. | scnb=2.0 |
| readexclusions yes | Read exclusion lists from the PARM7 file. | default in amber |
| cutoff 9 | Cut-off for vdW and electrostatic interactions. | cut=9 |
| watermodel {tip3, tip4} | Expect water residues to be TIP3P or TIP4P type. | detected automatically |
| pairListDist 11 | Distance between pairs of atoms for inclusion in pair lists that are used to calculate vdW and electrostatic interactions. | skinnb=2.0 |
| LJcorrection on | Apply an analytical tail correction to the reported vdW energy and virial that is equal to the amount lost due to switching and cutoff of the LJ potential. | vdwmeth=1 |
| ZeroMomentum on | Remove center of mass drift due to PME. | netfrc = 1 |
| rigidBonds {water, all} | SHAKE bonds that contain H atoms from water residues or all residues. | ntc=2, ntf=2, noshakemask = ... |
| rigidTolerance 1.0e-8 | SHAKE tolerance. | tol=1.0e-8 |
| rigidIterations 100 | SHAKE maximum iterations. | N/A |
| useSettle {on} | Use faster SETTLE instead of SHAKE for bonds belonging to water molecules. Default option in namd is on. | jfastw=0 |
| timeStep 1.0 | Integration time step in fs. | dt=0.001 |
| Non-bonded interactions | ||
| fullElectFrequency 1 | The number of timesteps between each full electrostatics evaluation. | N/A |
| nonBondedFreq 1 | How often short-range nonbonded interactions should be calculated. | N/A |
| stepspercycle 10 | Number of timesteps in each cycle. Each cycle represents the number of timesteps between atom reassignments to pair lists. | nbflag=0, nsnb=10 (although here I let amber do its default behavior) |
| PME on | Use PME. | always ON if ntb>0 |
|
PMEGridSizeX intx, PMEGridSizeY inty, PMEGridSizeZ intz |
The size of the fftw grid on each dimention. | nfft1=intx , nfft2=inty, nfft3=intz |
| PMETolerance 1.0e-6 | PME direct space tolerance. | dsum_tol=1.0e-6 |
| PMEInterpOrder 4 (i.e. cubic spline) | PME interpolation order. | order=4 |
|
cellBasisVector1 x1 y1 z1, cellBasisVector1 x2 y2 z2, cellBasisVector1 x3 y3 z3 |
The periodic cell vectors. | Defined indirectly at the bottom of thr .rst/.crd files. |
| cellOrigin x y z | Used as the center of the cell for wrapped output coordinates. | N/A |
|
Temperature Control - Thermostats [A] [A]
Note that PMEMD and NAMD provide more than the Langevin thermostat for pressure regulation. Sander and PMEMD provide the Andersen thermostat. NAMD implements the Lowe-Andersen thermostat. Please consult the users manuals for more details.
|
||
| langevin {on,off} | Use Langevin thermostat. | ntt=3 |
| langevinDamping 5 | Damping coefficient for Langevin dynamics in ps − 1. | gamma_ln = 5 |
| langevinTemp 300 | Langevin thermostat target temperature in K. | temp0 = 300 |
| langevinHydrogen off | Turn off the Langevin thermostat coupling of hydrogens. | default in amber (?) |
| temperature 300 | If no initial velocities are provided, choose this as the inital temperature. | tempi = 300 |
|
Pressure Control - Barrostats [B] [B]
Note that NAMD users usually chose to use Nosé-Hoover Langevin barrostat instead of the Beredensen version.
|
||
| BerendsenPressure {on,off} | Use Beredensen barrostat. | ntp=1 |
| BerendsenPressureTarget 1.01325 | Target Pressure in bars. | pres0 = 1.01325 |
| BerendsenPressureRelaxationTime 100.0 | Relaxation time for the Beredensen barrostat in ps for PMEMD and fs for NAMD. | taup = [1.0,5.0] |
|
useGroupPressure yes (needed for rigidBonds), useFlexibleCell no useConstantArea no |
Isotropic pressure scaling. | set when ntp=1 |
| Minimization, Dynamics | ||
| minimize nsteps | Do minimization for nsteps. | imin=1, maxcyc = nsteps |
| run nsteps | Run dynamics. | imin =0, nstlim = nsteps |
3 Modifying COULOMB conversion factors
If you want to make careful comparisons of the energies in NAMD and Amber, you need to modify the NAMD code to use the same conversion factors, particularly for converting electrostatic interactions to kcal/mol. These constants depend on the program:
AMBER = 332.0522173 CHARMM = 332.054 NAMD = 332.0636
To make the change, edit line 44 of the src/common.h file in NAMD 2.7 source code to read
#define COULOMB 332.0522173
then recompile.
4 Using non-cubic periodic boxes
4.1 Truncated Octahedron (TO)
To use a TO in sander/pmemd you have to use the solvateOct command in tleap/sleapm, or to modify the last line of the crd/rst file to the corresponding values. NAMD will not read the box information from the crd/rst file generated from tleap/sleap. Instead you will have to specify the box information using the cellBasisVector1, 2, 3. If the d is the length of the edge of the TO, than the three vectors should be:
cellBasisVector1 d 0.0 0.0 cellBasisVector2 (-1/3)*d (2/3)sqrt(2)*d 0.0 cellBasisVector3 (-1/3)*d (-1/3)sqrt(2)*d (-1/3)sqrt(6)*d
The last line of your crd/rst file gnerated from tleap/sleap or from sander/pmemd should look like:
d d d 109.4712190 109.4712190 109.4712190
4.2 Rhombic dodecahedron (RHDO)
Similarly, for a RHDO NAMD should read:
cellBasisVector1 d 0.0 0.0 cellBasisVector2 (1/2)*d (1/2)sqrt(3)*d 0.0 cellBasisVector3 (1/2)*d (1/6)sqrt(3)*d (1/3)sqrt(6)*d
This settings will make the periodic box to be wrapped so that in the (x, y) plane you will see hexagon. There is another set of settings that will generate after wrapping a square in the (x, y) plane:
cellBasisVector1 d 0.0 0.0 cellBasisVector2 0.0 d 0.0 cellBasisVector3 (1/2)*d (1/2)*d (1/2)sqrt(2)*d
The settings that will have to be added to the bottom of the crd/rst file will be:
d d d 60.0 60.0 60.0 or d d d 60.0 60.0 90.0
Note that tleap/sleap do not have a custom command for RHDO. You will have to modify the crd/rst file by hand.
4.3 Hexagonal Prism (HexP)
For a hexagonal prism, NAMD should read:
cellBasisVector1 d 0.0 0.0 cellBasisVector2 (1/2)*d (1/2)sqrt(3)*d 0.0 cellBasisVector3 0.0 0.0 h
wher d is the length of the side of the hexagon and h is the height of the prism. The settings that will have to be added to the bottom of the crd/rst file will be:
d d h 60.0 90.0 90.0
Note that tleap/sleap do not have a custom command for HexP. You will have to modify the crd/rst file by hand.
5 Using TIP4P water models in NAMD
To use any TIP4P-like water model in NAMD, you should set to on the leap FlexibleWater parameter. For example, a leap session that uses TIP4P/Ew would look like:
source leaprc.ff10 WAT = T4E loadAmberParams frcmod.tip4pew set default FlexibleWater on x = loadpdb "SOURCE.pdb" saveamberparm x x.parm7 x.crd savepdb x x.pdb quit
In your NAMD input file you should set watermodel option to tip4.
6 Sample inputs and outputs
Running an MD simulation of in the NVE ensemble, using periodic boundary condition and PME.
6.1 PMEMD input
Typical Production MD NVE &cntrl ntx=1, irest=0, ntc=2, ntf=2, tol=1.0e-8, nstlim=1000, dt=0.001, ntpr=1, ntwx=1, ntwr=1, cut=9.0 ntt=0, ntb=1, ntp=0, ioutfm=1, ntave=1000 / &ewald dsum_tol = 1.0e-6 nfft1=72, nfft2=72, nfft3=72, order=4, /
6.2 NAMD input
## OUTPUT/INPUT # Amber/(t,s,x)leap generated parm and crd file parmfile prmtop ambercoor xyz # Input bincoordinates input.restart.coor binvelocities input.restart.vel extendedSystem input.restart.xsc # Output restartfreq 1000 dcdfreq 1000 xstFreq 1000 outputEnergies 1000 outputPressure 1000 outputname output temperature 0 ## SIMULATION PARAMETERS # AMBER FF settings amber on rigidBonds all useSettle on rigidTolerance 1.0e-8 cutoff 9.0 pairlistdist 11.0 switching off exclude scaled1-4 readexclusions yes 1-4scaling 0.83333333 scnb 2.0 zeromomentum on ljcorrection on watermodel tip3 # Integrator Parameters timestep 1.00 nonbondedFreq 1 fullElectFrequency 1 stepspercycle 10 # Constant Temperature Control is off langevin off # Constant Pressure Control is off langevinPiston off # PME settings PME on PMETolerance 1.0e-6 PMEInterpOrder 4 FFTWUseWisdom no PMEGridSizeX 54 PMEGridSizeY 54 PMEGridSizeZ 54 # periodic cell cellBasisVector1 50.8726134 0.0 0.0 cellBasisVector2 0.0 50.8903175 0.0 cellBasisVector3 0.0 0.0 50.8639184 cellOrigin 0.0 0.0 0.0 ## EXECUTION SCRIPT run 100000